- Рахитоподобные болезни
- — группа наследственных тубулопатий, клиническая картина которых в ранние сроки заболевания имитирует рахит, но не связана с дефицитом поступающего в организм витамина D; их ведущим синдромом являются аномалии скелета (почечные остеопатии)
Патогенетические механизмы формирования первичных (наследственных) тубулопатий связывают со следующими факторами: генетически детерминированными нарушениями структуры мембранных белков-носителей; энзимопатиями наследственно обусловленной недостаточностью ферментов, обеспечивающих активный мембранный транспорт (см. Ферментопатии); изменением чувствительности рецепторов клеток канальцевого эпителия к действию гормонов; изменениями общей структуры цитомембран клеток (см. Мембраны биологические) при дисплазиях, в происхождении которых определенная роль принадлежит наследственным факторам. Вторичные тубулопатий возникают в результате повреждения транспортных систем почечных канальцев как при наследственных, так и при приобретенных болезнях обмена в связи с нарушениями метаболизма за пределами нефрона. Они развиваются также при воспалительных заболеваниях почек, что может обусловить значительные дифференциально-диагностические затруднения.
К рахитоподобным болезням относятся витамин-D-резистентный рахит, витамин-D-зависимый рахит, болезнь де Тони — Дебре — Фанкони и почечный тубулярный ацидоз.
Витамин-D-резистентный рахит (семейный гипофосфатемический рахит, фосфат-диабет) характеризуется доминантным типом наследования, сцепленным с полом, возможен также аутосомно-доминантный тип (см. Наследственность). Патогенез метаболических расстройств при этом заболевании сложен и в достаточной степени не исследован. Развитие его связывают с первичным нарушением процессов всасывания кальция и фосфора в кишечнике; с первичным дефектом транспорта неорганических фосфатов в почках и повышением чувствительности эпителия канальцев почек к действию паратгормона; с генетически детерминированным сочетанием этих нарушений; с синтезом в организме фосфатурических метаболитов витамина D и недостаточным образованием 25-оксихолекальциферола в печени.
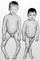
Заболевание проявляется у детей в 1—2 года, но может начаться в более старшем возрасте. Основными проявлениями болезни служат задержка роста и выраженные прогрессирующие деформации скелета, особенно нижних конечностей (по варусному типу, D-образные искривления; рис. 1, а), что сопровождается нарушением походки ребенка («утиная походка»); значительная болезненность костей и мышц, нередко мышечная гипотония; гипофосфатемия и гиперфосфатурия при нормальном содержании кальция в крови (дифференциально-диагностический признак); выявляемые рентгенологически рахитоподобные изменения костей, преимущественно нижних конечностей; сохранности интеллекта у больных детей.
Витамин-D-резистентный рахит отличается выраженным клиническим полиморфизмом. В зависимости от сроков манифестации, клинико-биохимических особенностей, чувствительности и характера ответной реакции на витамин D выделяют 4 клинико-биохимических варианта заболевания. Первый вариант характеризуется ранней (на первом году жизни) манифестацией, незначительной степенью костных деформаций, гипофосфатемией, гиперфосфатурией, повышением уровня паратгормона в крови, хорошей переносимостью витамина D. Второй вариант отличается более поздней (на втором году жизни) манифестацией, выраженными костными изменениями, гипофосфатемией, значительной гиперфосфатурией, резистентностью к высоким дозам витамина D. Третьему варианту присущи поздние сроки проявления заболевания (в 5—6-летнем возрасте), тяжесть поражений скелета, выраженная гипофосфатемия, значительное снижение абсорбции фосфора в кишечнике при нормальной или незначительной гиперфосфатурии; отмечается также нечувствительность к витамину D. Четвертый вариант характеризуется манифестацией на втором году жизни, умеренной степенью костных деформаций, повышенной чувствительностью к витамину D и склонностью к развитию клинико-биохимической картины гипервитаминоза D (рвота, тошнота, жажда, гиперкальциемия, гиперкальциурия и др.) в ответ на небольшие дозы витамина D. Клинический полиморфизм, особенности патогенеза и метаболических расстройств, широкий диапазон ответной реакции на витамин D свидетельствуют о генетической гетерогенности витамин-D-резистентного рахита.
Основными критериями диагностики витамин-D-резистентного рахита служат: клинические проявления — варусный тип и прогрессирующий характер костных деформаций нижних конечностей с отставанием физического развития; доминантный, сцепленный с Х-хромосомой тип наследования патологии; биохимические отклонения в обмене электролитов — низкий уровень фосфора в сыворотке крови, гиперфосфатурия, нормальные показатели общего кальция в крови (2,3—2,75 ммоль/л), повышение активности щелочной фосфатазы крови, повышение уровня паратгормона в крови, снижение абсорбции кальция и фосфора в кишечнике (см. Мальабсорбции синдром); результаты рентгенологического исследования, при котором выявляют два типа костных изменений: либо изменения эпифизов костей с расширением зон пролиферации хряща, грубоволокнистую структуру кости с признаками остеоидной гиперплазии (полная аналогия с рахитом), либо поражение метафизов уже сформировавшейся кости и проявления остеомаляции. Гистологически в костной ткани обнаруживают нарушение структуры костных каналов и трабекул, пролиферацию хряща, чередование усиленного образования остеоидной ткани с участками остеопороза.
Дифференциальный диагноз витамин-D-резистентного рахита и других рахитоподобных болезней представлен в таблице. Кроме того, дифференциальный диагноз проводят с рядом заболеваний наследственного и приобретенного характера — болезнью Бланта (при которой основным признаком является нарастающая варусная деформация большеберцовой кости как при отсутствии изменений в других отделах костно-суставной системы, так и метаболических расстройств с системными остеодисплазиями — фиброзной остеодисплазией, остеогенезом несовершенным, мраморной болезнью, Педжета болезнью и др.
Таблица
Дифференциальная диагностика рахитоподобных болезней у детей (по А.В. Шилову и П.В. Новикову)
|
Признаки |
Витамин-D-резистентный рахит |
Витамин-D-зависимый рахит |
Болезнь де Тони — Дебре — Фанкони |
Почечный тубулярный ацидоз |
|
Сроки манифестации |
1 год 3 мес. — 1 год 6 мес. |
3—5 мес. |
1—2 года |
5—6 мес. — 2—3 года |
|
Первые клинические проявления |
Варусные деформации нижних конечностей, рахитические «браслеты», «четки», нарушение походки |
Изменения со стороны ц.н.с. (раздражительность, нарушение сна, плаксивость), потливость, мышечная гипотония, снижение аппетита |
«Беспричинные» подъемы температуры, длительный субфебрилитет, полиурия, полидипсия, мышечные боли |
Полиурия, полидипсия, признаки функциональных нарушений нервной системы (раздражительность, плаксивость), резкая мышечная гипотония, мышечные боли |
|
Специфические признаки |
Прогрессирующий характер варусных деформаций нижних конечностей |
Выраженные костные изменения скелета: краниотабес, лобные и теменные бугры, рахитические «четки», деформации нижних конечностей; системный остеопороз |
Периодические подъемы температуры тела, прогрессирующие множественные костные деформации: увеличение печени, склонность к запорам |
Полиурия, полидипсия, мышечная гипотония, вплоть до адинамии; увеличение печени, запоры, прогрессирующие вальгусные деформации нижних конечностей |
|
Тип наследования |
Доминантный, сцепленный с X-хромосомой |
Аутосомно-рецессивный |
Аутосомно-рецессивный |
Аутосомно-рецессивный |
|
Особенности костной патологии |
Варусная деформация нижних конечностей; грубые бокаловидные деформации метафизов; искривления и утолщения трубчатых костей за счет одностороннего (чаще медиального) утолщения коркового слоя периоста |
Костные деформации (преимущественно нижних конечностей) различной степени выраженности; системный остеопороз |
Общее поражение костной ткани; выраженный системный остеопороз: трабекулярная исчерченность в дистальных и проксимальных отделах диафизов |
Вальгусная деформация нижних конечностей; системный остеопороз, смазанность, нечеткость контуров метафизов; ширина рахитической зоны до 2 см; нередко концентрическая атрофия кости |
|
Особенности состояния мочевыделительной системы |
Без изменений |
Без изменений |
Снижение аммониоацидогенетической функции почек; часто вторичный пиелонефрит |
Снижение ацидогенеза, вторичный пиелонефрит, нефрокальциноз |
|
Особенности состояния сердечно-сосудистой системы |
Без изменений |
Без изменений |
Артериальная гипотензия, изменения в миокарде (по ЭКГ) |
Артериальная гипотензия, изменения в миокарде вследствие электролитного дисбаланса |
|
Особенности физического развития |
Дефицит роста при неизмененной массе тела |
Отставание ростовесовых показателей |
Сочетания низкого роста и пониженного питания |
Сочетания низкого роста и резко пониженного питания |
|
Уровень психомоторного развития |
Нормальный |
Некоторая задержка моторного развития |
Нормальный |
Нормальный |
Лечение витамин-D-резистентного рахита как и других рахитоподобных болезней , должно быть комплексным. Оно направлено на коррекцию метаболических расстройств, профилактику осложнений и предупреждение инвалидизации ребенка. Показаниями для лекарственной терапии служат: активный процесс в костной ткани (по ренгенологическим данным), повышение активности щелочной фосфатазы крови, повышенная экскреция фосфатов с мочой, подготовка больных к хирургической коррекции. Основными препаратами в терапии витамин-D-резистентного рахита являются витамин и его метаболиты. Начальные дозы витамина D составляют 10 000 — 15 000 ЕД в сутки. Их увеличивают под контролем показателей кальция и фосфора в сыворотке крови и моче, активности щелочной фосфатазы крови, исследования уровня которых должно проводиться каждые 10—14 дней. Максимальные суточные дозы витамина D в зависимости от клинико-биохимических вариантов болезни составляют: при первом варианте 85 000—100 000 ЕД, при втором 150 000—200 000 ЕД, при третьем — 200 000—300 000 ЕД. При четвертом варианте болезни назначение витамина D противопоказано. Из метаболитов витамина D используют оксидевит в суточной дозе 0,25—3 мкг, при применении которого необходим особенно строгий контроль за уровнем кальция в крови (определяется 1 раз в 7—10 дней). В амбулаторных условиях исследуют мочу на содержание кальция путем постановки реакции Сульковича. Противопоказаниями для консервативной терапии витамином D и его метаболитами являются индивидуальная непереносимость препаратов, выраженная гиперкальциурия (более 4 ммоль/сут.), отсутствие активного процесса в костной ткани по данным лабораторных и рентгенологических исследований. В комплекс лечения витамин-D-резистентного рахита включают препараты кальция: глюконат кальция или хлорид кальция по 1,5—2 г в сутки) и фосфора (фитин 1—1,5 г в сутки, глицерофосфат кальция 0,5—1 г в сутки). Для улучшения процессов всасывания кальция и фосфора в кишечнике рекомендуют длительное (5—6 мес.) применение концентрированных цитратных смесей (например, лимонная кислота 24 г, цитрат натрия 48 г и дистиллированная вода 500 мл) по 20—50 мл в сутки. В активной фазе болезни при наличии болей в костях и суставах показан постельный режим продолжительностью до 2 нед. В период клинико-лабораторной ремиссии и наблюдения больных в амбулаторных условиях ограничивают физические нагрузки, занятия физическими упражнениями и лечебный массаж проводят по специальной щадящей программе, назначают соляно-хвойные ванны, осуществляют санаторно-курортное лечение.
Показателями эффективности консервативной терапии являются улучшение общего состояния больных, увеличение темпов роста детей, нормализация или значительное улучшение показателей фосфорно-кальциевого обмена, снижение активности щелочной фосфатазы крови и положительная динамика структурных изменений костной ткани (по данным рентгенологического исследования).
Хирургическое лечение витамин-D-резистентного рахита сводится к корригирующим остеотомиям костей голеней или бедренных костей с последующей иммобилизацией повязкой или дистракционно-компрессионным аппаратом. Обязательным условием для проведения хирургического лечения служит достижение стойкой клинико-биохимической ремиссии в течение не менее 2 лет.
Витамин-D-зависимый рахит (псевдовитамин-D-дефицитный рахит псевдорахит) является энзимопатией, которая связана с дефектом фермента 1-альфа-гидроксилазы в почках, осуществляющего превращение 25-оксихолекальциферола в 1,25-диоксихолекальциферол. Имеет аутосомно-рецессивный тип наследования, однако встречаются и спорадические случаи заболевания, обусловленные, видимо, свежими первичными мутациями.
Патогенез витамин-D-зависимого рахита можно представить следующим образом: дефицит 1-альфа-гидроксилазы почек ® недостаточный синтез 1,25-диоксихолекальциферола ® снижение абсорбции кальция в кишечнике ® гипокальциемия ® вторичный гиперпаратиреоз ® нарушение фосфорно-кальциевого обмена ® развитие рахитоподобных изменений скелета.
Клинически патология выявляется чаще всего в первые 3—5 мес. жизни ребенка (реже болезнь начинается в 3—5-летнем возрасте). В начальных стадиях характеризуется функциональными изменениями ц.н.с. и вегетативной нервной системы (потливость, нарушение сна, вздрагивания и др.), к которым позднее присоединяются костные поражения. Последние отличаются прогредиентностью, несмотря на ранее проведенную профилактику рахита или проводимое обычное антирахитическое лечение. В зависимости от глубины метаболических расстройств выделяют два клинико-биохимических варианта витамин-D-зависимого рахита: с тяжелой и умеренной степенью выраженности обменных нарушений и костных деформаций. Для первого варианта болезни характерны серьезные костные изменения (варусные, варусно-саблевидные деформации нижних конечностей, деформации грудной клетки, черепа, предплечий, рахитические «четки», «браслетки»; рис. 2), выраженная гипокальциемия (1,4—1,7 ммоль/л), высокие показатели активности щелочной фосфатазы крови, нормальный или слегка сниженный уровень фосфатов в крови, повышенная экскреция фосфатов и значительное уменьшение выделения кальция с мочой, генерализованная гипераминоацидурия. Рентгенологически определяют глубокие нарушения структуры костной ткани (генерализованный остеопороз, широкая рахитическая зона, неровность контуров метафизов и др.). Второму варианту свойственны легкие или умеренные деформации костей преимущественно нижних конечностей с негрубыми структурными изменениями костной ткани по рентгенологическим данным, умеренная гипокальциемия (1,9—2,2 ммоль/л). Развитие первого варианта патологии связывают с выраженным дефицитом 1,25-диоксихолекальциферола, второго — со снижением чувствительности органов-мишеней к этому метаболиту витамина D.
Основными критериями диагностики служат клинические проявления, семейный характер заболевания с аутосомно-рецессивным типом наследования, отмеченные особенности метаболических сдвигов и отсутствие эффекта от проводимого антирахитического лечения витаминов D. Наибольшие дифференциально-диагностические затруднения возникают при разграничении витамин-D-зависимого и витамин-D-дефицитного рахита. В пользу первого свидетельствуют прогрессирующий характер костных деформаций и отсутствие признаков нормализации клинико-биохимических показателей при контрольном лечении витамином D в дозе 4 000 ЕД в сутки в течение 6—8 нед., а также низкий уровень кальция и нормальное содержание 25-оксихолекальциферола в крови.
Особенностью лечения витамин-D зависимого рахита является необходимость заместительной терапии оксидевитом — аналогом биологически активного метаболита витамина D 1,25-диоксихолекальциферола, суточные дозы которого составляют 0,5—4 мкг в зависимости от индивидуальной переносимости препарата и тяжести течения заболевания. Используют также витамин D в дозах от 10 000—15 000 ЕД до 40 000—60 000 ЕД в сутки. В комплекс лечебных средств обязательно рекомендуют включать препараты кальция и фосфора, витамина А, С, Е, цитратные смеси курсами по 3—5 мес. Положительная динамика показателей фосфорно-кальциевого гомеостаза наблюдается обычно через 4—6 нед. после начала комплексной терапии. После отмены препаратов витамина D может развиться (чаще через 3—6 мес.) рецидив заболевания, поэтому лечение следует проводить непрерывно в течение нескольких лет. При рано назначенной и адекватной терапии клинико-биохимические признаки болезни у детей раннего возраста подвергаются обратному развитию. В случаях поздней диагностики, когда уже имеются тяжелые и грубые деформации нижних конечностей, затрудняющие передвижение больных, показана корригирующая остеотомия, условием успешного проведения которой является стойкая клинико-биохимическая ремиссия в течение 11/2—-2 лет.
Болезнь де Тони — Дебре — Фанкони (глюкоаминофосфат-диабет) представляет собой наиболее тяжелое заболевание среди рахитоподобных болезней . Наследуется по аутосомно-рецессивному типу, однако экспрессивность мутантного гена в гомозиготном состоянии значительно варьирует; встречаются спорадические случаи, обусловленные свежей мутацией.
Полагают, что в основе болезни лежат генетически обусловленные дефекты ферментативного фосфорилирования в почечных канальцах (комбинированная тубулопатия). Это приводит к нарушению процессов энергообеспечения транспорта фосфатов, глюкозы и аминокислот в почечных канальцах и повышенной их экскреции с мочой, а также расстройству механизмов поддержания равновесия кислот и оснований. Развивающийся метаболический ацидоз и недостаток фосфорных соединений способствуют нарушению формирования костной ткани по типу остеомаляции и рахитоподобных изменений скелета. В ряде случаев выявляют морфологические изменения в почечных канальцах, нарушение функции паращитовидных желез, расстройства синтеза 1,25-диоксихолекальциферола.
В большинстве случаев первые признаки заболевания появляются во второй половине первого года жизни, развернутый симптомокомплекс формируется ко второму году жизни; реже наблюдается поздняя манифестация болезни — в 6—7-летнем возрасте. Начальные клинические проявления — повышенная жажда, полиурия, иногда длительный субфебрилитет, рвота. На втором году жизни выявляют отставание физического развития и костные деформации нижних конечностей (вальгусные или варусные), грудной клетки, предплечий и плечевых костей (рис. 3). Рентгенологически при этом определяют остеопороз системный различной степени выраженности, истончение коркового слоя трубчатых костей, разрыхление зон роста, отставание темпов роста костной ткани от биологического возраста ребенка.
Характерными особенностями биохимических нарушений при болезни де Тони — Дебре — Фанкони являются снижение уровня кальция и фосфора в крови, повышение активности щелочной фосфатазы, метаболический ацидоз (рН — 7,35—7,25; ВЕ = —10—12 ммоль/л). Экскреция кальция с мочой обычно остается нормальной при повышенном клиренсе фосфатов мочи. Отмечают глюкозурию (20—30 г/л и выше), генерализованную гипераминоацидурию и нарушение функций аммониоацидогенеза — снижение титрационной кислотности, повышение рН мочи > 6,0. При полиурии до 2 л и более в сутки удельная плотность мочи, как правило, высокая (1025—1035), что обычно связано с глюкозурией.
В зависимости от тяжести клинических проявлений и метаболических расстройств выделяют два клинико-биохимических варианта болезни де Тони — Дебре — Фанкони. Первый характеризуется значительной задержкой физического развития, тяжелым течением заболевания с выраженными костными деформациями и нередко переломами костей, резкой гипокальциемией (1,6—1,8 ммоль/л), снижением абсорбции кальция в кишечнике. При втором варианте отмечают умеренную задержку физического развития, легкое течение с незначительными костными деформациями, нормокальциемию и нормальное усвоение кальция в кишечнике.
Критериями диагностики являются выраженный дефицит массы тела и роста ребенка, задержка становления статико-моторных функций, рахитоподобные деформации скелета с характерной рентгенологической картиной нарушений структуры костной ткани, отмеченные особенности электролитных нарушений.
Дифференциальный диагноз проводят с рахитом, остеопатиями вследствие хронической почечной недостаточности (см. Остеопатия нефрогенная); кроме того, большое значение имеет дифференцирование первичной болезни де Тони — Дебре — Фанкони с вторичным синдромом, обнаруживаемым при других наследственных и приобретенных заболеваниях (синдроме Лоу, ювенильном нефронофтизе, цистинозе, тирозинемии, галактоземии, гликогенозах, наследственной непереносимости фруктозы, гепатоцеребральной дистрофии, миеломной болезни, амилоидозе, синдроме Шегрена, нефротическом синдроме, при почечной трансплантации, гиперпаратиреозе, поражении почек солями тяжелых металлов, отравлении лекарственными веществами, в т. ч. витамином D, лизолом и т.д.). Синдром Лоу (окулоцереброренальный синдром) в отличие от болезни де Тони — Дебре — Фанкони характеризуется отставанием в умственном развитии, двусторонней катарактой, глаукомой, гипорефлексией, а также рецессивным, связанным с полом типом наследования. Ювенильный нефронофтиз Фанкони, морфологической основой которого являются кисты в мозговом веществе почек на уровне собирательных трубочек и гиалиноз почечных клубочков, отличается ранним нарушением концентрационной функции почек (гипостенурией), нормохромной анемией, гиперазотемией; рахитоподобные изменения скелета присоединяются позже. Решающую роль в диагностике нефронофтиза Фанкони играет исследование биоптата почечной ткани.
Основные принципы лечения заключаются в коррекции электролитных нарушений, сдвигов в кислотно-щелочном равновесии, устранении дефицита калия и бикарбонатов. Особенности применения витамина D и его метаболитов для ликвидации нарушений фосфорно-кальциевого гомеостаза сводятся к необходимости проведения лечения повторными курсами (начальная суточная доза витамина D 25 000—30 000 ЕД, максимальная — 75 000—150 000 ЕД, доза оксидевита 0,5—1,5 мкг в сутки), т.к. при отмене препаратов часто наблюдаются рецидивы (так называемые метаболические кризы, прогрессирование остеопороза и рахитоподобных изменений костной ткани). В комплекс лечения включают препараты кальция, фосфора, витамины А, С, Е, группы В в возрастных дозах. Показано ограничение поваренной соли и включение в рацион продуктов, оказывающих ощелачивающее действие, а также богатых калием. В фазе ремиссии назначают массаж, соляно-хвойные ванны. Хирургическая коррекция при болезни де Тони — Дебре — Фанкони целесообразна только при развитии тяжелых костных деформаций и достижении стойкой клинико-биохимической ремиссии в течение 2 лет.
Почечный тубулярный ацидоз в широком понимании — клинический синдром, характеризующийся постоянным метаболическим ацидозом, низким уровнем бикарбонатов и увеличенной концентрацией хлора в сыворотке крови. У большинства больных постоянно выявляется гипокалиемия из-за потери калия с мочой, сопровождающаяся мышечной слабостью вплоть до параличей; частыми проявлениями патологии служат остеомаляция, нефрокальциноз и мочекаменная болезнь, которые связаны с сопутствующими нарушениями фосфорно-кальциевого обмена. Как синдром почечный тубулярный ацидоз развивается при многих заболеваниях и состояниях, таких как пиелонефрит, интерстициальный нефрит с папиллитом, хроническая почечная недостаточность, нефрокальциноз различного происхождения, гипервитаминоз D, иммунные заболевания почек, гликогенозы, синдром Лоу, отравления солями тяжелых металлов и др.
Почечный тубулярный ацидоз рассматривается и как самостоятельная нозологическая форма, наследственное заболевание, называемое болезнью Лайтвуда — Баттлера — Олбрайта (синдром Лайтвуда — Олбрайта). Первый («классический») тип болезни обусловлен дефектом ацидогенетической функции дистальных почечных канальцев; второй тип зависит от дефекта проксимальных канальцев, не способных реабсорбировать бикарбонаты при сохранности ацидогенетической функции дистальных канальцев. Почечный тубулярный ацидоз наследуется по аутосомно-рецессивному типу, но не исключаются аутосомно-доминантное наследование и спорадические случаи, обусловленные первичными мутациями.
Первые признаки болезни появляются на первом году жизни: отмечаются снижение аппетита, полиурия, полидипсия, быстрая утомляемость. Дети начинают рано отставать в физическом развитии. На втором году жизни развиваются деформации скелета (вальгусные деформации нижних конечностей, «четки», «браслеты», лобные и теменные бугры), выраженная мышечная гипотония. К 2 годам формируется полный симптомокомплекс почечного тубулярного ацидоза. Иногда наблюдается поздняя манифестация (в 5—6-летнем возрасте) в виде прогрессирующих деформаций нижних конечностей (рис. 1, б) и нарушение походки. Рентгенологические изменения костной ткани не имеют специфических особенностей и напоминают таковые при болезни де Тони — Дебре — Фанкони.
Почечному тубулярному ацидозу свойственны следующие биохимические нарушения: метаболический ацидоз (рН — 7,35—7,25, дефицит ВЕ = —10—20 ммоль/л), гипофосфатемия, гипокальциемия, повышение активности щелочной фосфатазы, низкий уровень экскреции титруемых кислот с мочой и повышенный до 7,1 рН мочи, значительное снижение концентрационной функции почек (относительная плотность мочи 1001—1008). В ряде случаев наблюдаются умеренные протеинурия и лейкоцитурия. При рентгенологическом и ультразвуковом исследовании мочевой системы примерно у половины больных выявляют множественные конкременты, расположенные не только в собирательной системе, но и в паренхиме почек (нефролитиаз и нефрокальциноз).
В зависимости от тяжести течения заболевания и глубины метаболических расстройств выделяют два клинико-биохимических варианта почечного тубулярного ацидоза, патогенетически отличающиеся степенью нарушения абсорбции кальция в кишечнике. При первом варианте наблюдаются значительная задержка физического развития, тяжелое течение заболевания с выраженными костными деформациями, гипокальциемия, гипокалиемия, значительный вторичный гиперпаратиреоидизм, сниженное усвоение кальция в кишечнике. Второй вариант характеризуется умеренной задержкой физического развития, относительно легким течением заболевания с незначительными костными деформациями, нормокальциемией и нормокалиемией, а также нормальным усвоением кальция в кишечнике.
Основными критериями диагностики почечного тубулярного ацидоза являются: раннее начало заболевания, задержка физическое развития, вальгусные деформации нижних конечностей, выраженный метаболический ацидоз, высокий рН мочи, гиперкальциурия, нефрокальциноз.
Дифференциальный диагноз проводят с заболеваниями, при которых почечный тубулярный ацидоз может быть лишь симптомом основной патологии. В этом случае диагностика базируется на учете признаков ведущего заболевания.
Особенностью лечения почечного тубулярного ацидоза является направленность на коррекцию метаболического ацидоза, гипокалиемии с помощью бикарбоната натрия, препаратов калия, цитратных смесей, димефосфона. При явлениях остеопороза и остеомаляции показано назначение препаратов витамина D или его метаболитов (начальные суточные дозы витамина D 10 000—20 000 ЕД, максимальные — 30 000—60 000 ЕД; суточные дозы оксидевита 0,5—2 мкг), препаратов кальция до нормализации его уровня в крови. При оксалатно-кальциевом нефролитиазе рекомендуют окись магния по 0,2—0,25 г в сутки курсами по 3—4 нед. в течение длительного времени, 15% раствор димефосфона в суточной дозе 1 мл на 5 кг массы тела, а также исключение из рациона продуктов, богатых оксалатами (щавель, шпинат, томатный сок, шоколад и др.). Лекарственную терапию проводят на фоне общеукрепляющего лечения с включением щелочных минеральных вод, фруктовых соков, ограничении белков животного происхождения, использованием комплекса витаминов (А, Е, группы В).
Под влиянием комплексного лечения у больных улучшаются общее состояние, показатели фосфорно-кальциевого обмена, активность щелочной фосфатазы крови, определяется положительная динамика рентгенологической картины структурных изменений костной ткани, особый контроль проводят за показателями кислотно-основного состояния (дефицит ВЕ), рН мочи, уровнем кальция и фосфора в крови, определение которых должно осуществляться 1 раз в 7—10 дней.
Хирургическое лечение рекомендуют только детям с выраженными костными деформациями нижних конечностей, которые затрудняют их передвижение, при этом необходима двухгодичная стабилизация клинико-биохимических показателей.
Библиогр.: Барашнев Ю.И. и Вельтищев Ю.Е. Наследственные болезни обмена веществ у детей, Л., 1978; Барашнев Ю.И., Руссу Г.С. и Казанцева Л.З. Дифференциальный диагноз врожденных и наследственных заболеваний у детей, Кишинев, 1984; Врожденные и наследственные заболевания у детей, под ред. Ю.И. Барашнева, с. 166, М., 1985; Игнатова М.С. и Вельтищев Ю.Е. Детская нефрология, Л., 1989; Кон Р.М. и Рот К.С. Ранняя диагностика болезней обмена веществ, пер. с англ., М., 1986; Почки и гомеостаз в норме и при патологии, под ред. С. Клара, пер. с англ., М., 1987.